NetSciReg'15 - Network Models in Cellular Regulation
June 1, 2015 - Zaragoza, Spain
| Synopsis |
| Program |
| NetSciReg'15 Flyer |
| Important Dates |
| Registration |
| Call for Contributed Talks |
| Logistics |
| NetSci 2015 |
| NetSciReg'14 |
| NetSciReg'13 |
| Sponsors |
|
Time: 1:10 - 1:30 PM
Type: Contributed presentation
Affiliation: Computer Laboratory, University of Cambridge
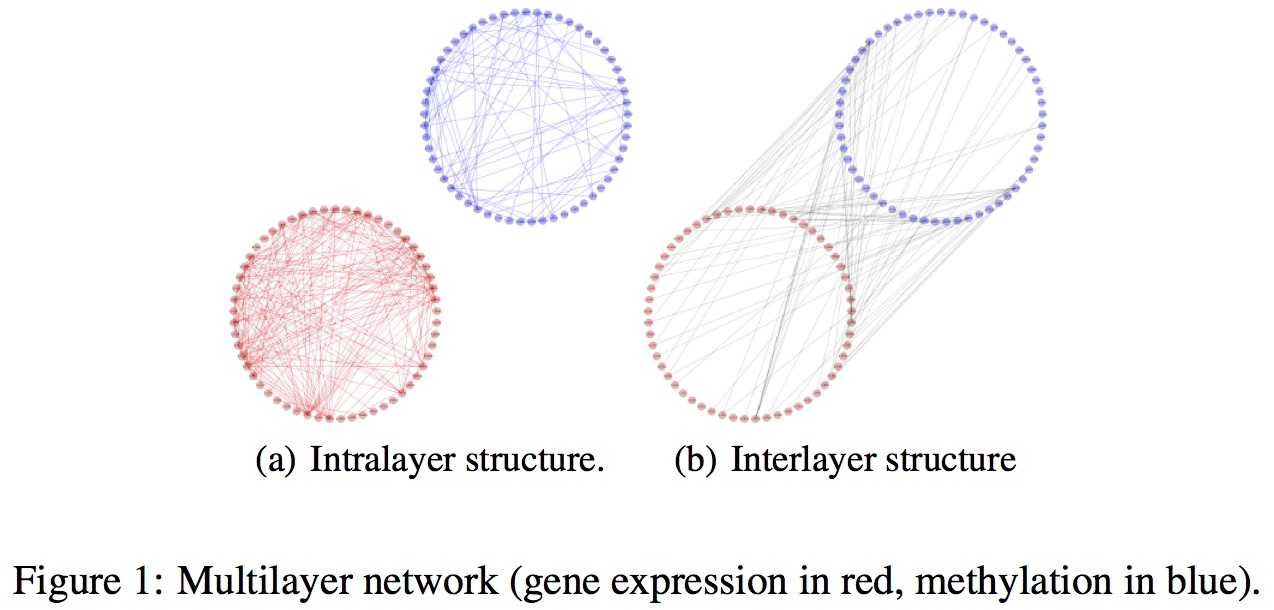
Abstract In the last two decades the emergence of high-throughput technologies has made available huge amounts of high-dimensional multi-omic data sets, revealing the multifaceted nature of most biological processes in living organisms [1]. Despite this abundance of data, our comprehension of structural patterns, functional prin- ciples, and systemic behaviours in complex systems is still approximate. A clear example of this gap between richness in data and poverty in knowledge is the difficulty in uncovering the relational structure between molecules within a living cell [2]. Genes, for instance, interact in a complex web of relations that or- chestrate various functions in response to both endogenous and exogenous stimuli. Traditional methods to reverse-engineering gene networks from measured expres- sion data include graphical Gaussian modeling (GGM) [3], Bayesian networks [4], relevance networks [5], and Pearson correlation networks [6], just to name a few. In this paper we propose a new approach to infer and study gene networks, com- bining methylation and gene expression data in a multilayer network [7, 8]. In- stead of using standard co-expression measures, such as Pearson's correlation co- efficient, mutual information, or Spearman's rank correlation coefficient, we build a multilayer network (Figure 1) computing a pairwise similarity between genes within each layer and between the layers by means of a Gaussian kernel. In par- ticular, we associate to each gene a feature vector consisting of three elements: the mean value of its expression/methylation level for healthy subjects, the mean value for patients, and a normalized value resulting from the t-test for the two populations (healthy subjects and patients). Our formulation implicitly embodies a plurality of relationships and functional roles between genes, making it possible to elucidate the dynamical mechanism of aggregation and disruption of connectivity structures across multiple omic layers. We analyze the role played by methylation in regulating gene expression for ten inflammatory diseases by considering traditional descriptive measures at both lo- cal and global scale and providing a Bayesian nonparametric generative model based to investigate the formation of mesoscopic structures.
References
[1] A. R. Joyce and B. O. Palsson, The model organism as a system: integrating 'omics' data sets,
Nature Reviews Molecular Cell Biology, vol. 7, pp. 198-210, Mar 2006. |
SPONSORS:
|